snpArcher
A Snakemake workflow for whole-genome resequencing and variant calling, designed for non-model organisms.
snpArcher takes you from raw FASTQ reads (or SRA accessions) to a filtered, analysis-ready VCF. It handles trimming, alignment, variant calling, joint genotyping, quality control, and postprocessing in a single reproducible pipeline. Built for researchers working with non-model organisms (birds, fish, insects, plants, and everything else without a curated reference panel).
What does snpArcher do?¶
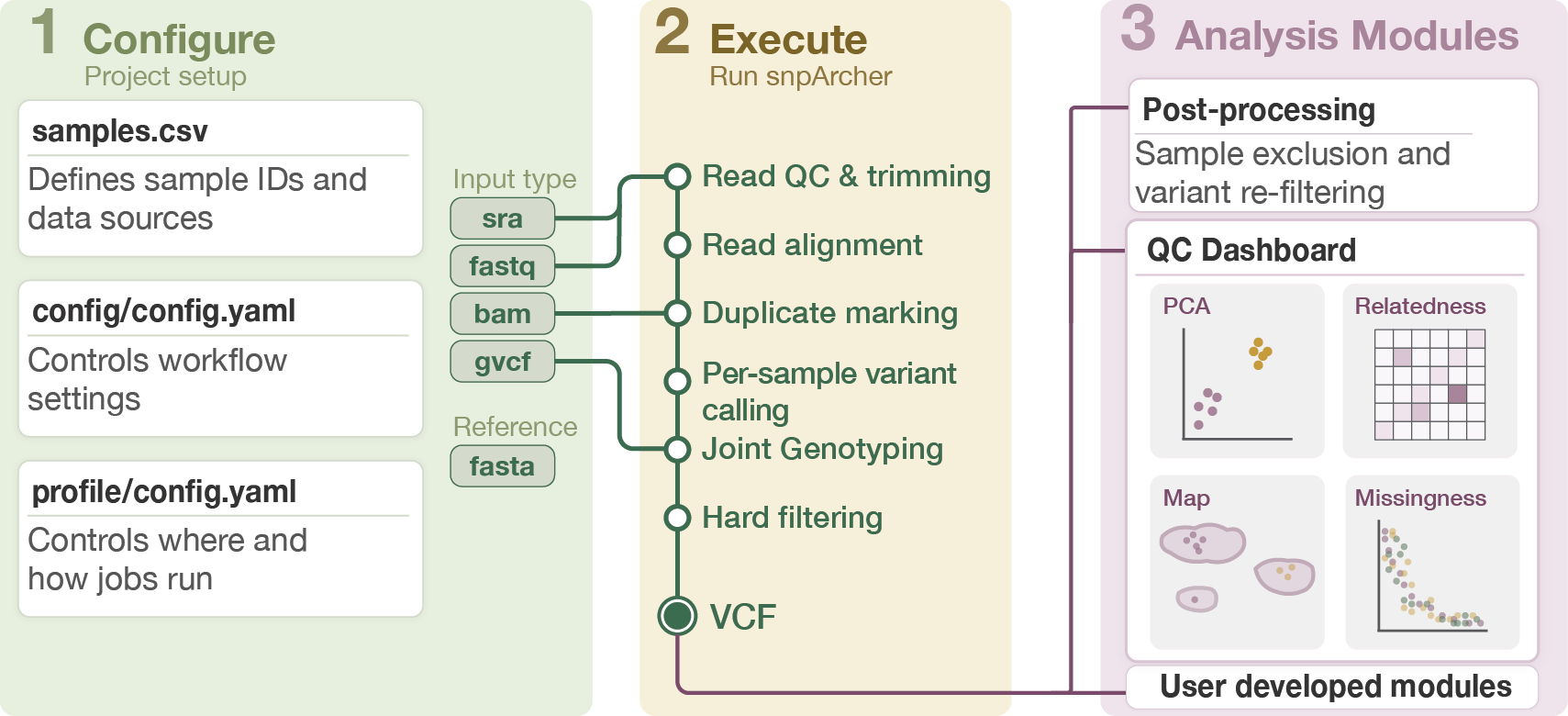
Plus optional modules for QC dashboards and postprocessing/filtering.
Where to start¶
| I want to… | Go to… |
|---|---|
| Install snpArcher | Installation guide |
| Verify my install works | Quickstart |
| Process my own data end-to-end | Your first project |
| Look up a config option | Config schema |
| Understand the pipeline design | Architecture |
Citing snpArcher¶
If you use snpArcher in your research, please cite:
Mirchandani, C.D. et al. (2024). A fast, reproducible, high-throughput variant calling workflow for population genomics. Molecular Biology and Evolution, 41(1), msad270. doi:10.1093/molbev/msad270